# get libraries
library(readr)
library(ggplot2)
library(dplyr)
library(tidyr)
library(stringr)
# VEP output in
vep_data <- read_tsv(
"variants/chr6ch17_mutect2_annotated_with_clinVar.txt",
comment = "##"
)
# better to set up a ggplot2 theme
theme_course <- function() {
theme_minimal(base_size = 14) +
theme(
plot.title = element_text(size = 16, face = "bold"),
axis.title = element_text(size = 14, face = "bold"),
axis.text = element_text(size = 12),
legend.title = element_text(size = 13, face = "bold"),
legend.text = element_text(size = 12),
strip.text = element_text(size = 13, face = "bold"),
legend.position = "bottom"
)
}
# Split when it contains multiple values
consequences_df <- vep_data |>
separate_rows(Consequence, sep = "&") |>
select(Consequence)
# get chr
vep_data <- vep_data |>
separate_wider_delim(Location, delim = ":", names = c("chr", "position")) |>
mutate(chr = factor(chr, levels = c("chr6", "chr17")))
# Extract IMPACT from Extra column
vep_data$IMPACT <- gsub(".*IMPACT=([^;]+).*", "\\1", vep_data$Extra)
# get nice data out of the df
extract_field <- function(data, field) {
gsub(paste0(".*", field, "=([^;]+).*"), "\\1", data$Extra)
}
# combine multiple fields
vep_data <- vep_data |>
mutate(
IMPACT = extract_field(pick(everything()), "IMPACT"),
SYMBOL = extract_field(pick(everything()), "SYMBOL"),
VARIANT_CLASS = extract_field(pick(everything()), "VARIANT_CLASS"),
BIOTYPE = extract_field(pick(everything()), "BIOTYPE")
)Day 3: Variant Analysis in R
Introduction
Now that we have annotated our somatic variants with VEP, we can move from command-line annotation to R-based analysis and visualization. In this section, we will load VEP output into R, explore the distribution of variant consequences, and create informative visualizations to help prioritize variants of interest.
Setup
ImportantSwitch to RStudio
For the R exercises, open RStudio in your browser. Use the same IP address as your VS Code environment, but change the port: replace 9xxx with 10xxx (e.g., if your VS Code is at http://12.345.678.91:9001, RStudio is at http://12.345.678.91:10001). Log in with username rstudio and the password provided.
First, we load the VEP annotated output into R and extract some useful fields from the Extra column. Copy and run this setup code in your RStudio console:
NoteSetup code
Exercise 5
ImportantVisualize the data from VEP
Objective: Learn how to quickly visualize your variants.
Task: Visualize:
- Consequence plot, to visualize the distribution of consequences?
- Consequence plot, by chromosome?
- Can we see the consequence colored by impact?
- Can we see the consequence colored by impact by chromosome?
- What are the most common variant classes?
- What are the most common biotypes?
- Bonus: ClinVar, SIFT, and PolyPhen
- Bonus: Gene level damage
Tip1. Consequence plot, to visualize the distribution of consequences
# consequence plot
ggplot(vep_data, aes(x = reorder(Consequence, table(Consequence)[Consequence]))) +
geom_bar() +
coord_flip() +
theme_minimal() +
labs(title = "Distribution of \n Variant Consequences",
x = "Consequence Type",
y = "Count") +
theme_course()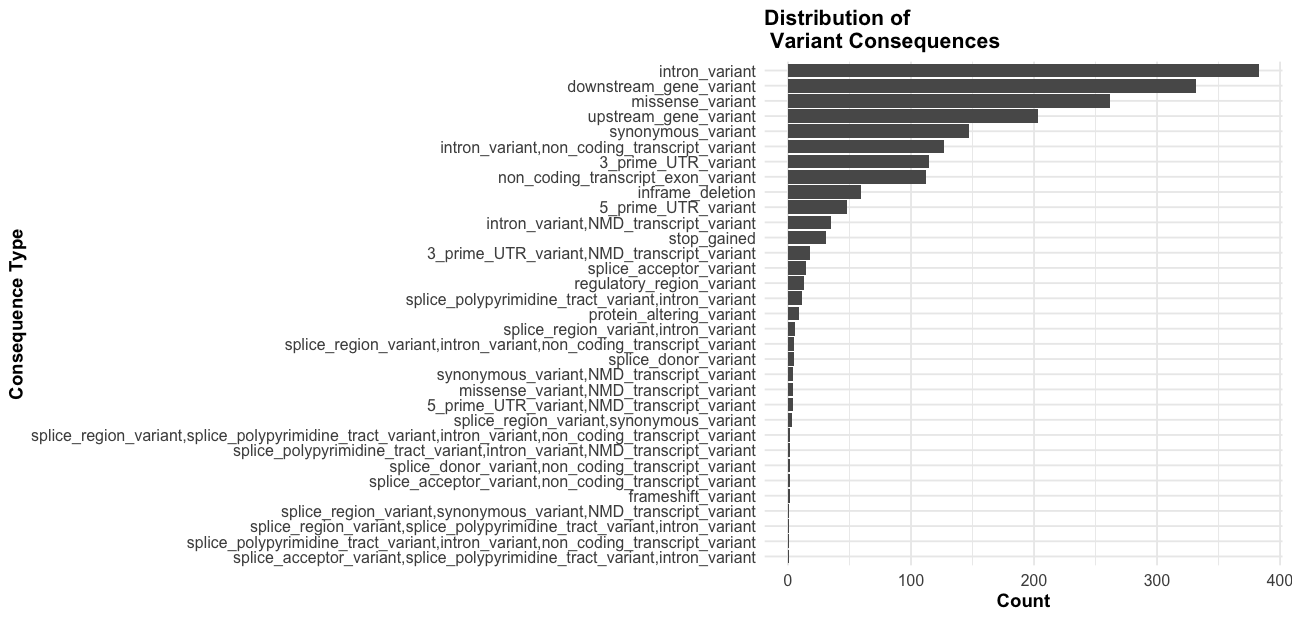
Tip2. Consequence plot, by chromosome
# consequence plot
ggplot(vep_data, aes(x = reorder(Consequence, table(Consequence)[Consequence]))) +
geom_bar() +
coord_flip() +
theme_minimal() +
labs(title = "Distribution of \n Variant Consequences",
x = "Consequence Type",
y = "Count") +
facet_grid(~chr) +
theme(
legend.position = "bottom"
) +
theme_course()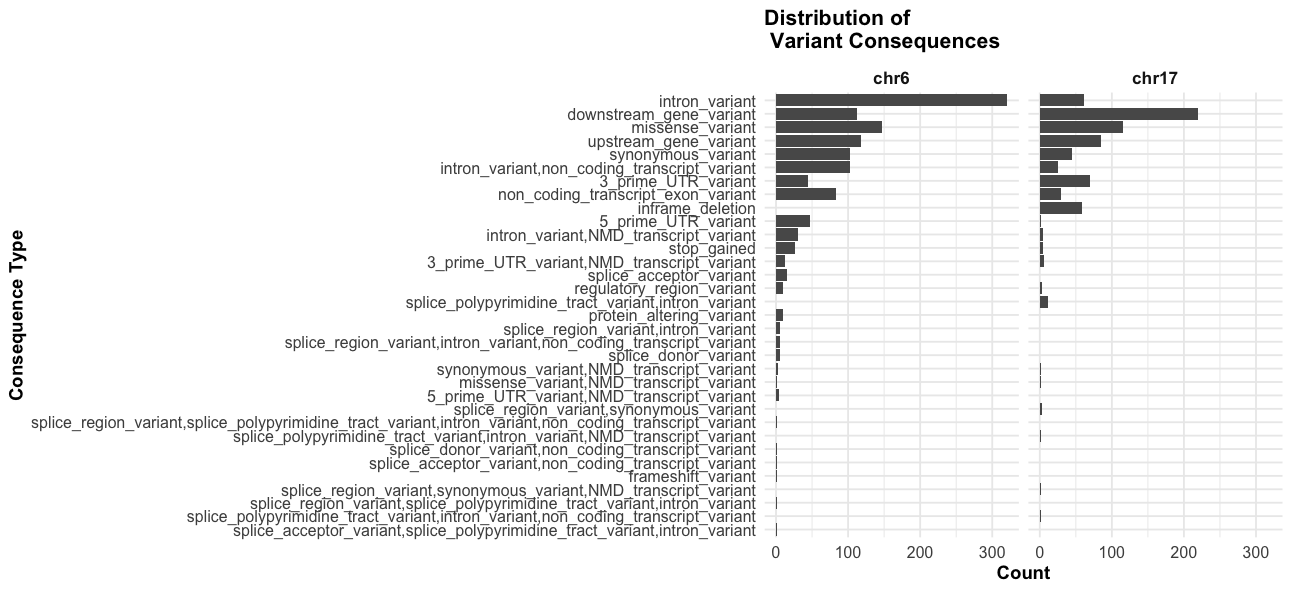
Tip3. Can we see the consequence colored by impact
# Impact + Consequence plot
ggplot(vep_data, aes(x = reorder(Consequence, table(Consequence)[Consequence]), fill = IMPACT)) +
geom_bar() +
coord_flip() +
theme_minimal() +
scale_fill_brewer(palette = "Set1") +
labs(title = "Variant Consequences \n by Impact",
x = "Consequence Type",
y = "Count") +
theme(
legend.position = "bottom"
) +
theme_course()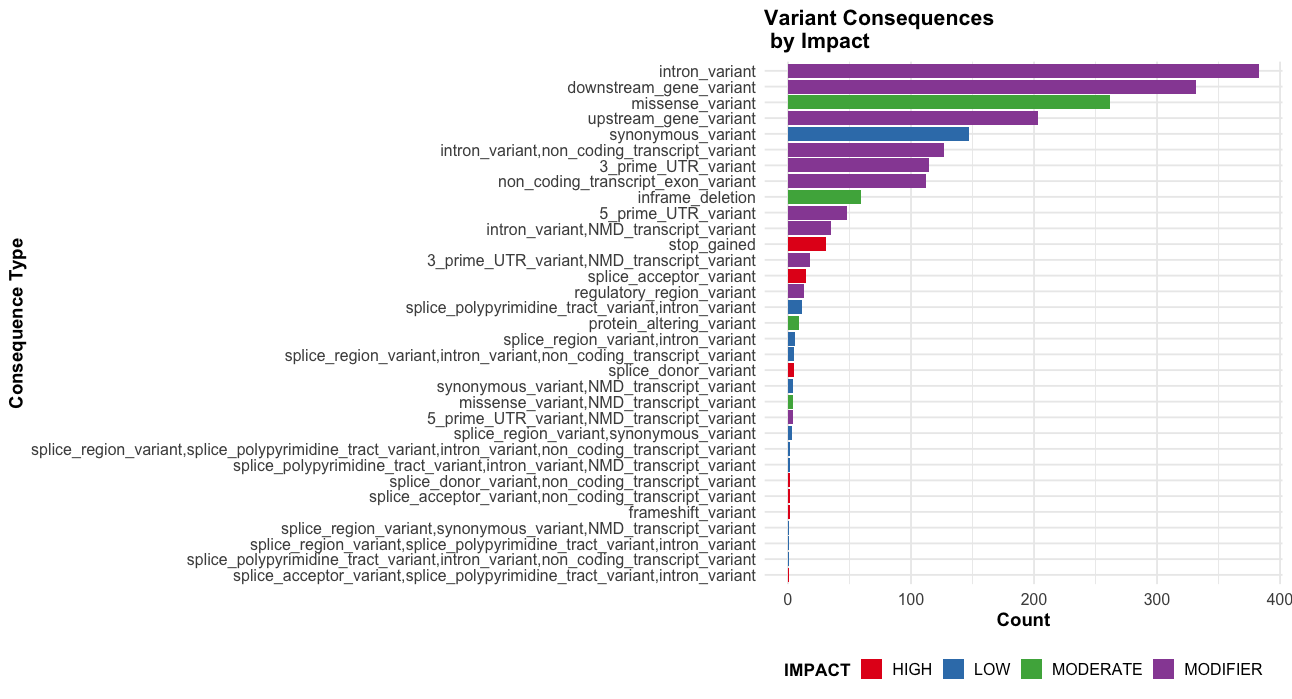
Tip4. Can we see the consequence colored by impact by chromosome
# Impact + consequence plot x chr
ggplot(vep_data, aes(x = reorder(Consequence, table(Consequence)[Consequence]), fill = IMPACT)) +
geom_bar() +
coord_flip() +
theme_minimal() +
scale_fill_brewer(palette = "Set1") +
labs(title = "Variant Consequences \n by Impact",
x = "Consequence Type",
y = "Count") +
facet_grid(~chr) +
theme(
legend.position = "bottom"
) +
theme_course()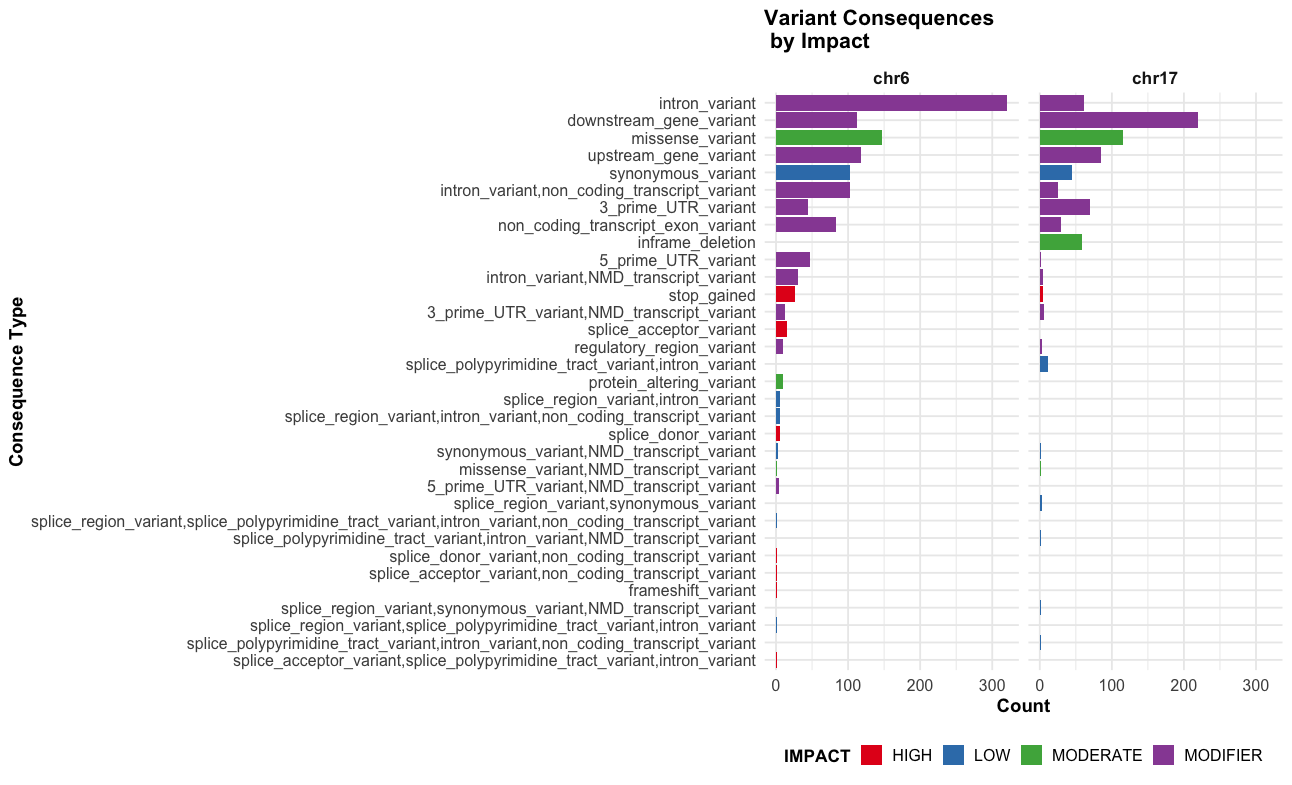
Tip5. What are the most common variant classes?
# variant class plot
vep_data |>
count(VARIANT_CLASS) |>
arrange(desc(n)) |>
ggplot(aes(x = reorder(VARIANT_CLASS, n), y = n)) +
geom_bar(stat = "identity", fill = "steelblue") +
coord_flip() +
theme_minimal() +
labs(title = "Distribution of \n Variant Classes",
x = "Variant Class",
y = "Count"
) +
theme_course()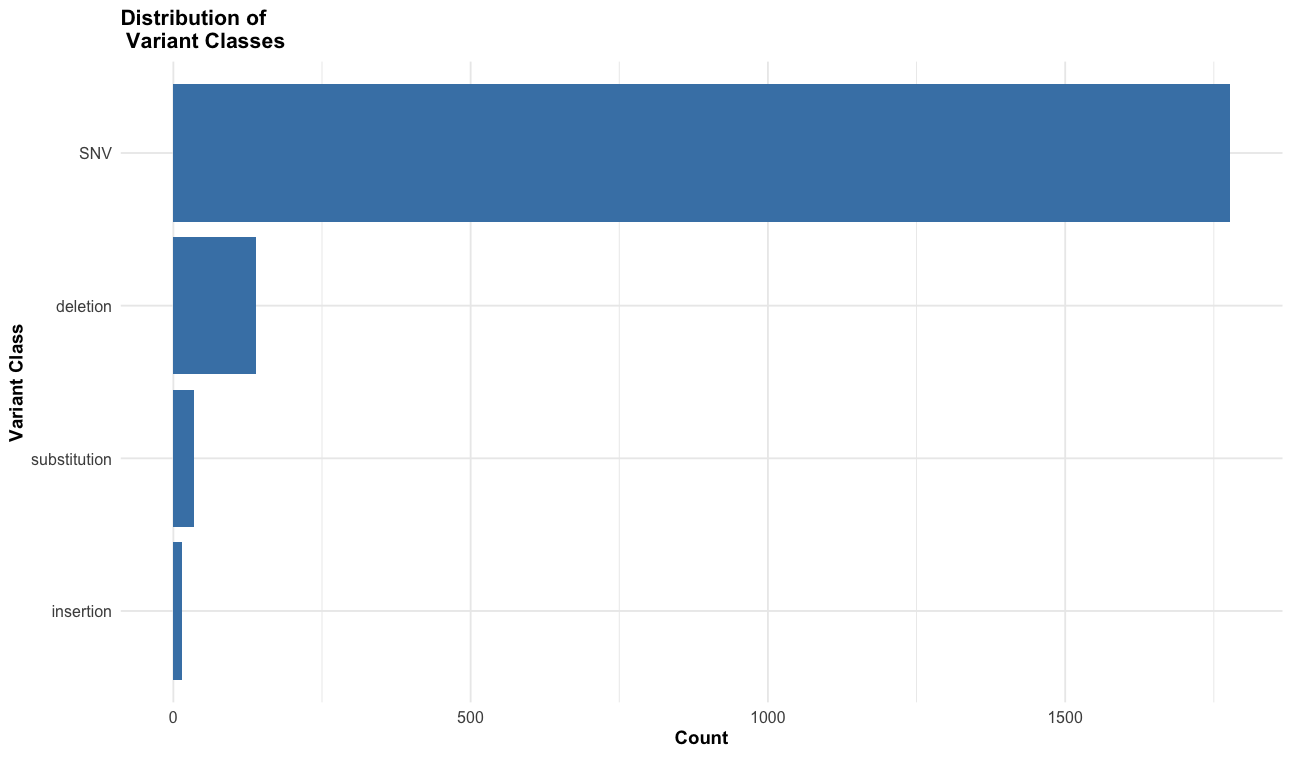
Tip6. What are the most common biotypes?
# biotype distribution
ggplot(vep_data, aes(x = reorder(BIOTYPE, table(BIOTYPE)[BIOTYPE]))) +
geom_bar(fill = "steelblue") +
coord_flip() +
theme_minimal() +
labs(title = "Distribution of \n Biotypes",
x = "Biotype",
y = "Count"
) +
theme_course()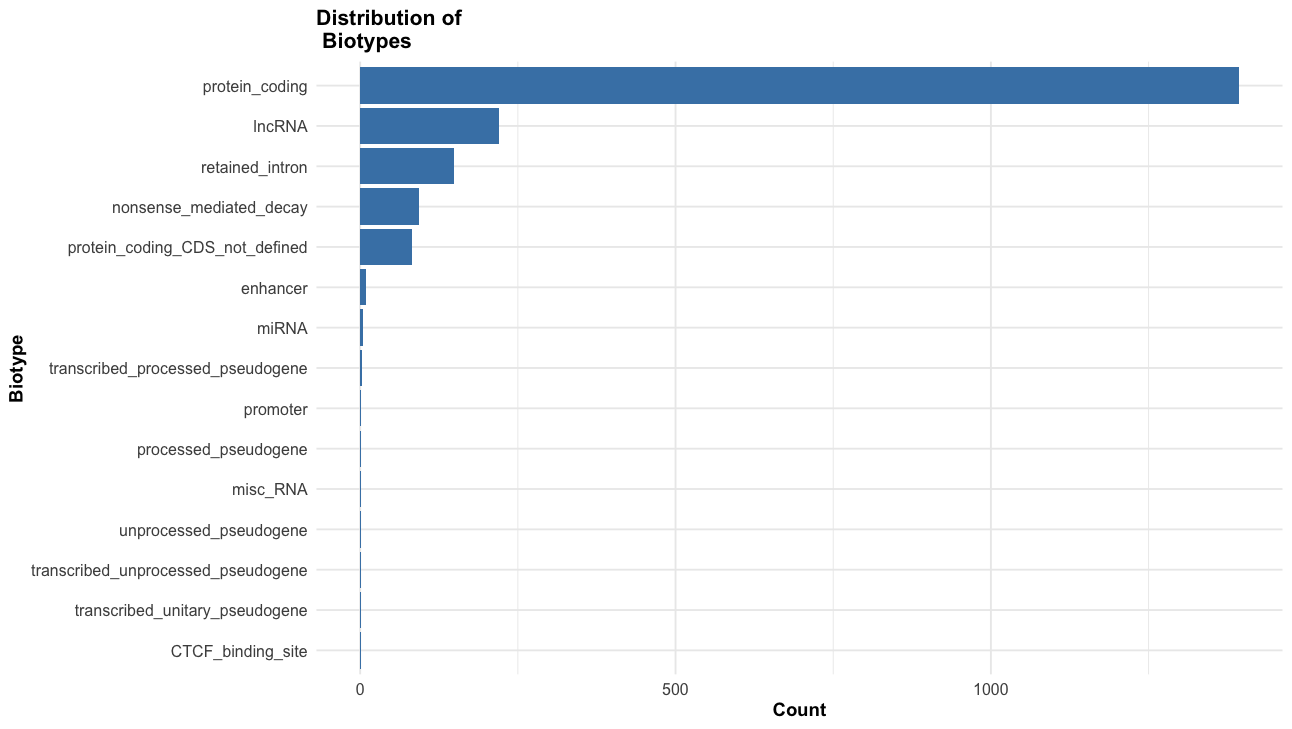
Tip7. Bonus: ClinVar, SIFT, and PolyPhen
# Extract SIFT
vep_data$sift <- str_extract(vep_data$Extra, "SIFT=.*?;")
vep_data$sift <- str_replace(vep_data$sift, "SIFT=", "")
vep_data$sift <- str_replace(vep_data$sift, ";", "")
vep_data$sift <- str_replace(vep_data$sift, "\\(.*\\)", "")
# Extract PolyPhen
vep_data$polyphen <- vep_data$Extra |>
str_extract("PolyPhen=.*?;") |>
str_replace("PolyPhen=", "") |>
str_replace(";", "") |>
str_replace("\\(.*\\)", "")
# Extract ClinVar
vep_data$clinvar <- vep_data$Extra |>
str_extract("ClinVar_CLNSIG=.*?;") |>
str_replace("ClinVar_CLNSIG=", "") |>
str_replace(";", "")
# Distribution of SIFT predictions
ggplot(subset(vep_data, !is.na(sift)),
aes(x = reorder(sift, table(sift)[sift]))) +
geom_bar(fill = "steelblue") +
coord_flip() +
theme_minimal() +
labs(title = "Distribution of SIFT Predictions",
x = "SIFT Prediction",
y = "Count",
caption = "Only missense variants are scored by SIFT") +
theme_course()
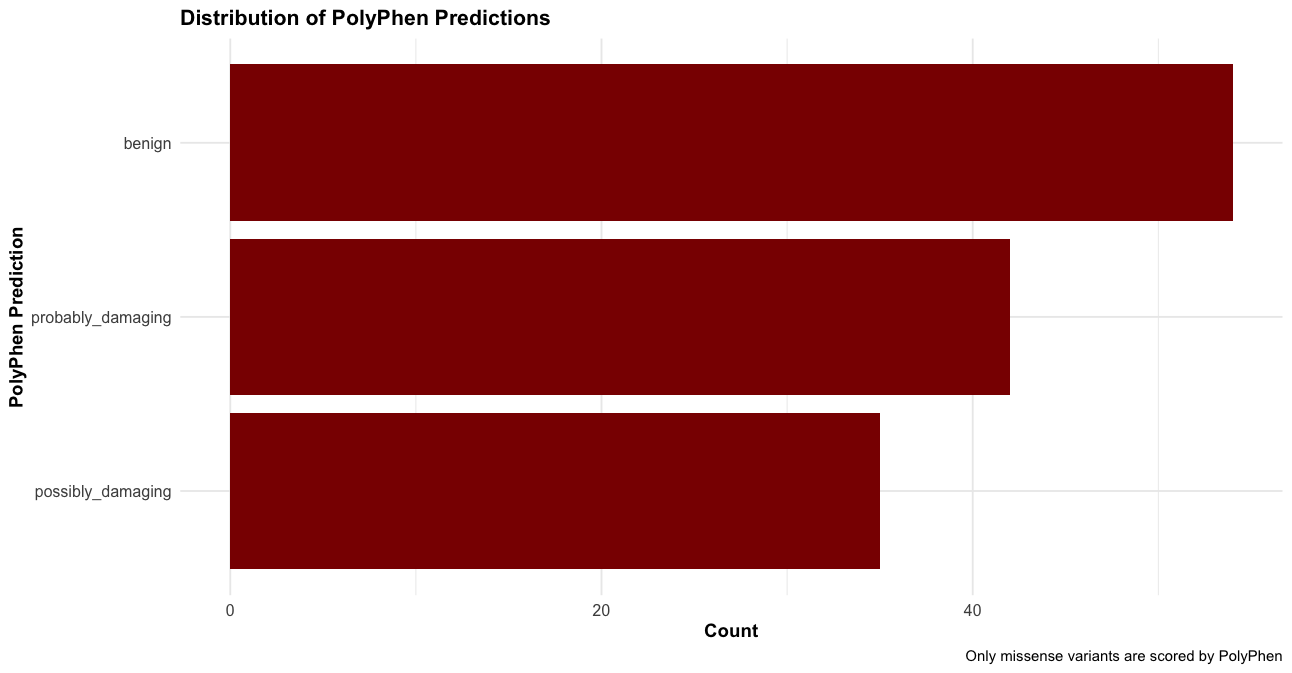
# Distribution of PolyPhen predictions
ggplot(subset(vep_data, !is.na(polyphen)),
aes(x = reorder(polyphen, table(polyphen)[polyphen]))) +
geom_bar(fill = "darkred") +
coord_flip() +
theme_minimal() +
labs(title = "Distribution of PolyPhen Predictions",
x = "PolyPhen Prediction",
y = "Count",
caption = "Only missense variants are scored by PolyPhen") +
theme_course()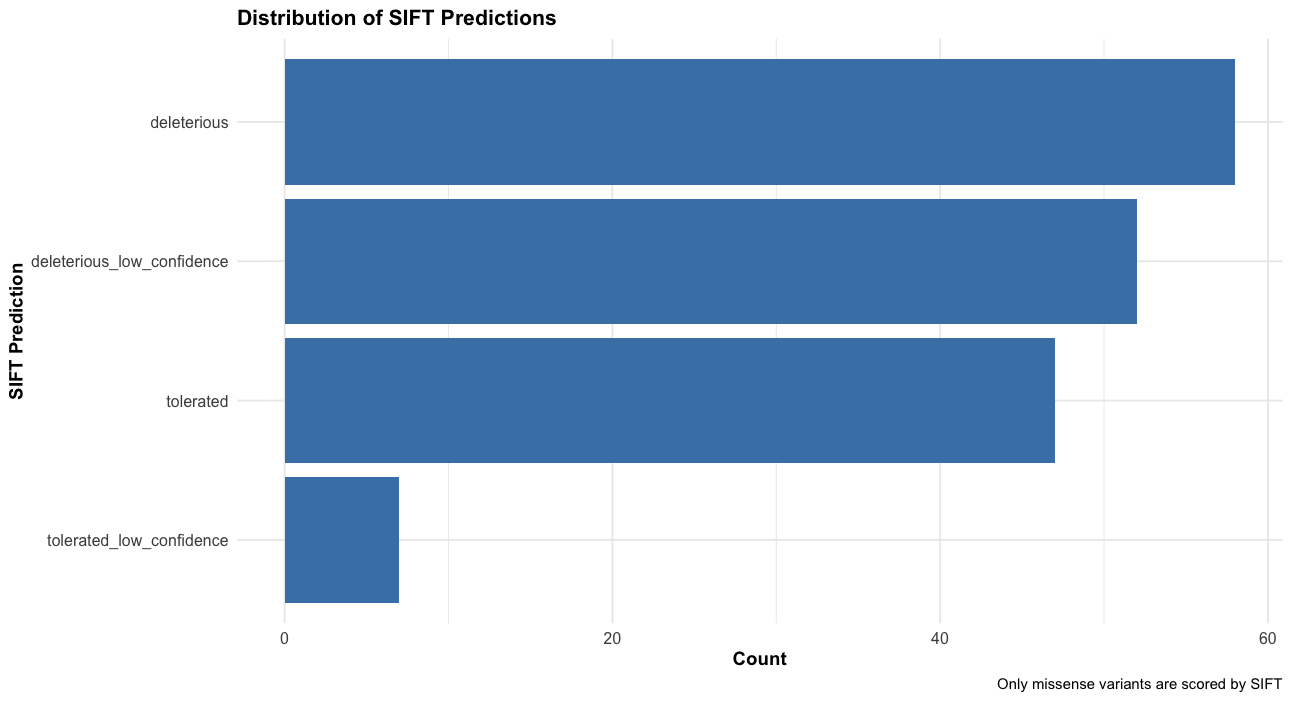
Combined SIFT and PolyPhen predictions with Impact
# Only for variants with both predictions
p3 <- subset(vep_data, !is.na(sift) & !is.na(polyphen)) |>
ggplot(aes(x = sift, fill = IMPACT)) +
geom_bar() +
facet_wrap(~polyphen) +
coord_flip() +
theme_minimal() +
scale_fill_brewer(palette = "Set1") +
labs(title = "SIFT and PolyPhen Predictions by Impact",
x = "SIFT Prediction",
y = "Count") +
theme(legend.position = "bottom") +
theme_course()
# 4. ClinVar significance with impact
p4 <- subset(vep_data, !is.na(clinvar)) |>
ggplot(aes(x = reorder(clinvar, table(clinvar)[clinvar]), fill = IMPACT)) +
geom_bar() +
coord_flip() +
scale_fill_brewer(palette = "Set1") +
theme_minimal() +
labs(title = "Clinical Significance by Impact",
x = "ClinVar Clinical Significance",
y = "Count") +
theme(legend.position = "bottom") +
theme_course()
# 5. For missense variants only - comparison of predictions
p5 <- subset(vep_data, Consequence == "missense_variant" & !is.na(sift) & !is.na(polyphen)) |>
ggplot(aes(x = sift, fill = polyphen)) +
geom_bar(position = "dodge") +
theme_minimal() +
scale_fill_brewer(palette = "Set2") +
labs(title = "SIFT vs PolyPhen Predictions\nfor Missense Variants",
x = "SIFT Prediction",
y = "Count",
fill = "PolyPhen Prediction") +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
theme_course()Plots
print(p3)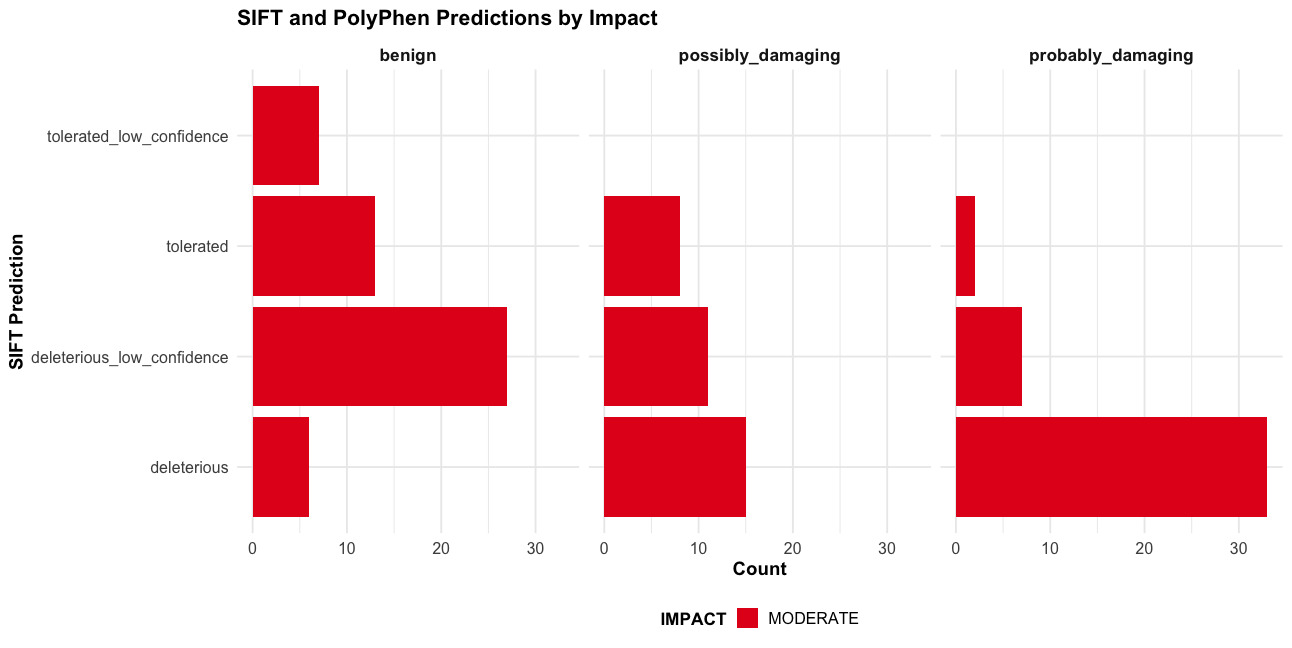
print(p4)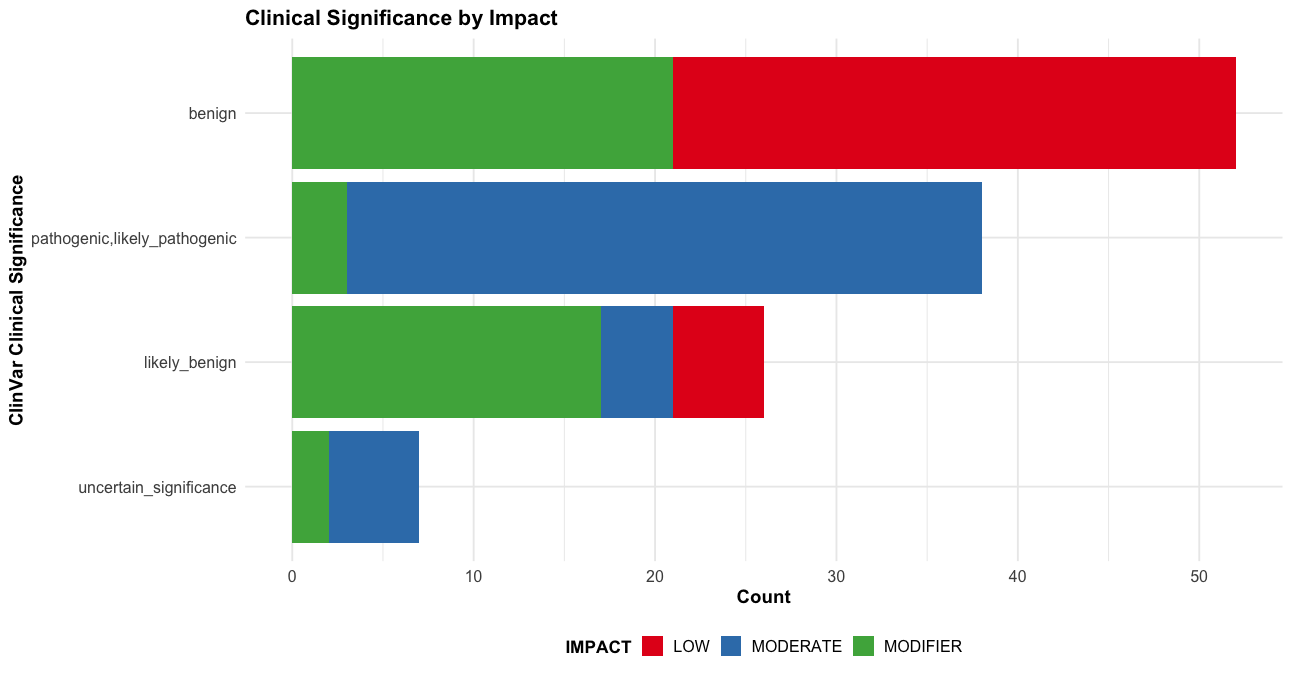
print(p5)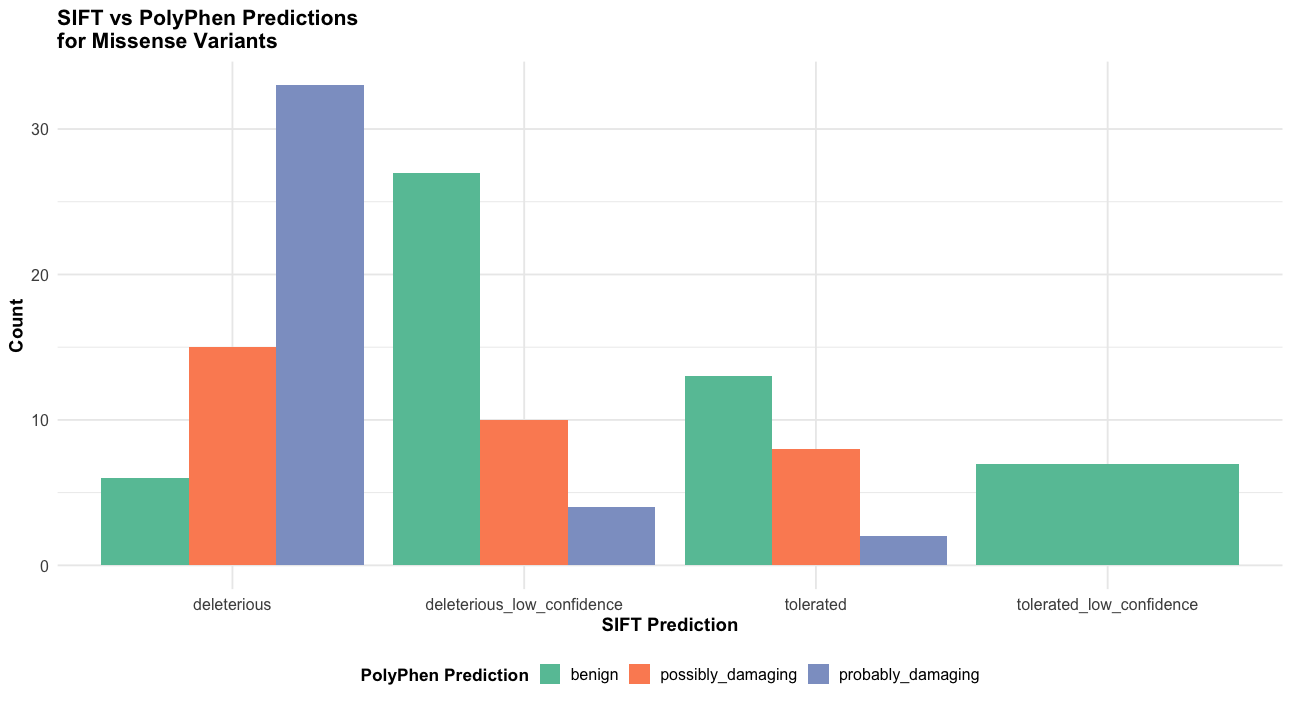
Tip8. Bonus: Gene level damage
# Gene-level summary - most frequently mutated genes
p6 <- vep_data |>
filter(!is.na(SYMBOL) & SYMBOL != "") |>
count(SYMBOL, chr) |>
arrange(desc(n)) |>
slice_head(n = 20) |>
ggplot(aes(x = reorder(SYMBOL, n), y = n, fill = chr)) +
geom_col() +
coord_flip() +
scale_fill_brewer(palette = "Set1") +
labs(title = "Top 20 Most Frequently Mutated Genes",
x = "Gene Symbol",
y = "Number of Variants",
fill = "Chromosome") +
theme_course()
# High impact variants by gene
p7 <- vep_data |>
filter(IMPACT %in% c("HIGH", "MODERATE") & !is.na(SYMBOL) & SYMBOL != "") |>
count(SYMBOL, IMPACT) |>
arrange(desc(n)) |>
slice_head(n = 30) |>
ggplot(aes(x = reorder(SYMBOL, n), y = n, fill = IMPACT)) +
geom_col() +
coord_flip() +
scale_fill_manual(values = c("HIGH" = "#d62728", "MODERATE" = "#ff7f0e")) +
labs(title = "Genes with High/Moderate Impact Variants",
x = "Gene Symbol",
y = "Number of Variants",
fill = "Impact Level") +
theme_course()Plots
print(p6)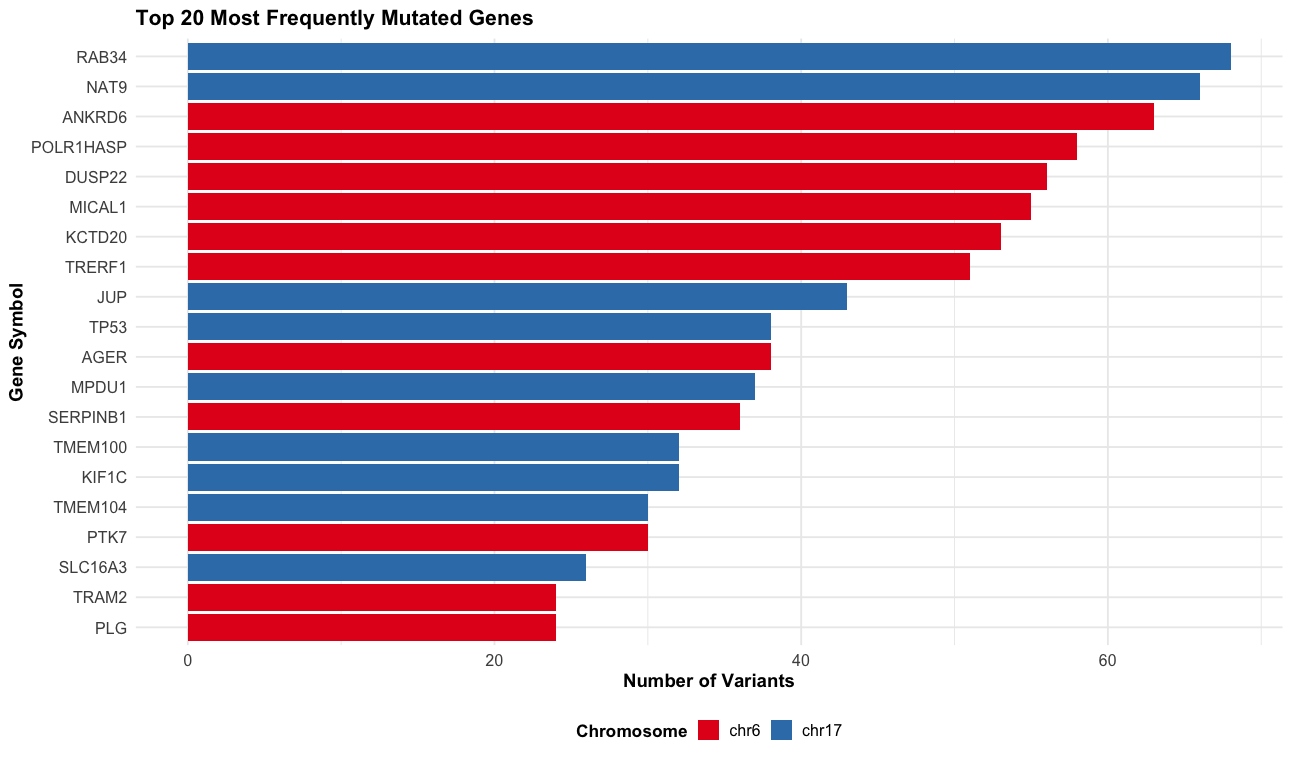
print(p7)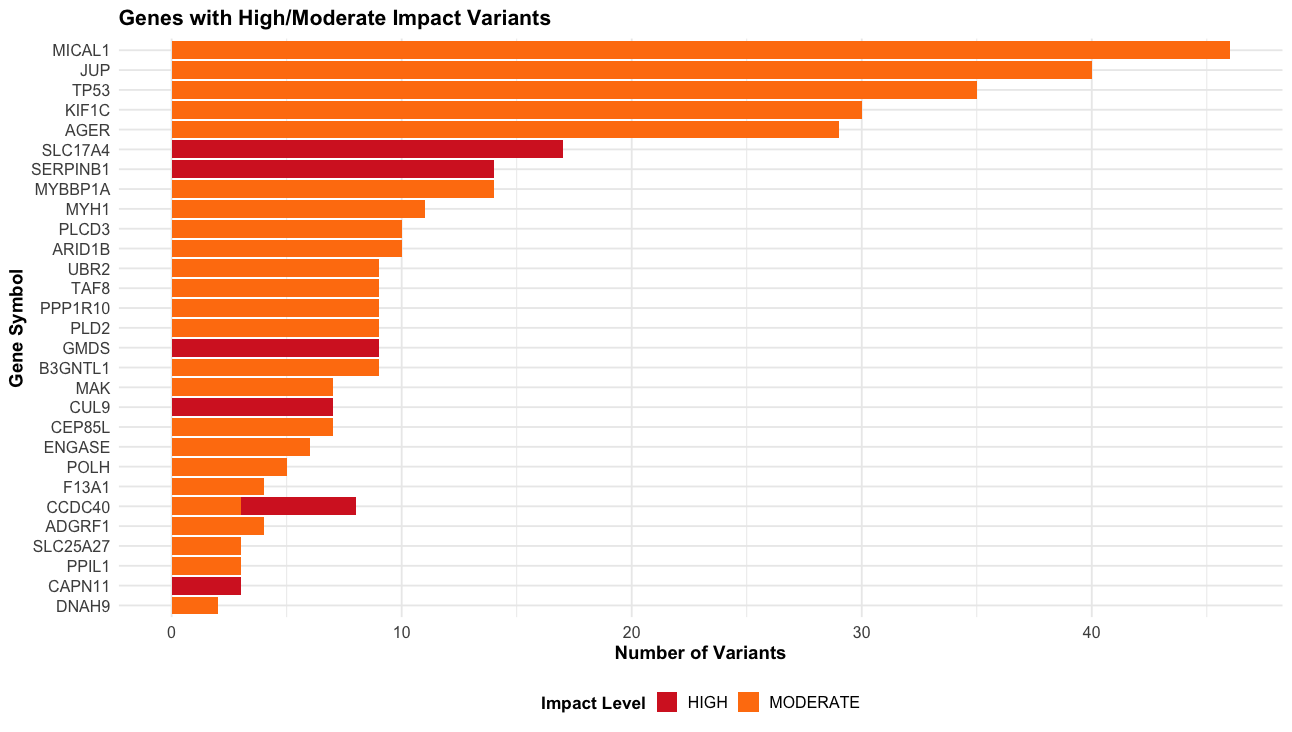
VEP Performance Optimization
For large-scale analyses, consider these optimization strategies:
- Using Cache:
--cache
--dir_cache /path/to/cache
--offline- Parallel Processing:
--fork 4- Minimal Output:
--fields "Consequence,IMPACT,SYMBOL,Feature"
--no_statsTips for Cancer Variant Analysis
- Quality Control
- Filter low-quality variants before VEP annotation
- Use matched normal samples when available (if not consider PONs)
- Consider sequencing artifacts
- Annotation Strategy
- Use multiple prediction algorithms
- Consider tissue-specific expression
- Include population frequencies
- Add clinical annotations
- Interpretation Guidelines
- Use cancer-specific frameworks for somatic variants
- Document all filtering criteria
- Maintain version control of annotation sources
Databases
Download data to COSMIC
- For COSMIC: We cannot provide those files (they require licensing), you need to register and download the files.
CosmicMutantExport.tsv.gzYou can register here:
Best Practices from Literature
NoteKey Recommendations
From McLaren et al. (2016) - VEP Paper:
- Use
--pickor--per_geneto manage transcript complexity - Combine multiple prediction algorithms for consensus
- Always document VEP version and cache version for reproducibility
For Cancer Analysis:
- Filter common variants (gnomAD MAF < 0.001, i.e., 0.1%) for somatic analysis — variants above this frequency are almost certainly germline
- Consider tissue-specific expression data when available
- Use multiple evidence sources (ClinVar + COSMIC + literature)
- Validate high-priority variants with orthogonal methods
Tools and Documentation
- VEP Documentation: http://www.ensembl.org/info/docs/tools/vep/
- VEP GitHub: https://github.com/Ensembl/ensembl-vep
- Cancer Gene Census: https://cancer.sanger.ac.uk/census
Remember to:
- Always verify file paths and permissions
- Check input data format and quality
- Validate output at each step
- Consider computational resources when processing large datasets
- Document any modifications to the pipeline
Quick Reference
TipVEP Command Cheatsheet
Basic annotation:
vep -i input.vcf \
--cache ${HOME}/project/data/.vep \
--offline \
--assembly GRCh38 \
-o output.txtWith common options:
vep -i input.vcf \
--cache ${HOME}/project/data/.vep \
--offline \
--assembly GRCh38 \
--everything \
--fork 2 \
--vcf \
-o output.vcfFilter high-impact:
filter_vep -i input.txt \
-o filtered.txt \
--filter "IMPACT is HIGH or IMPACT is MODERATE"Output format options:
# VEP default format (custom tab-delimited, suitable for filter_vep)
vep ... -o output.txt
# Explicit tab format with header (better for loading into R/pandas)
vep ... --tab -o output.txt
# VCF output (standard variant format)
vep ... --vcf -o output.vcf
# Compressed VCF (saves space, ready for tabix)
vep ... --vcf --compress_output bgzip -o output.vcf.gz
# Custom tab-delimited fields (choose specific columns)
vep ... --tab --fields "Location,Allele,Gene,SYMBOL,Consequence,IMPACT,SIFT,PolyPhen" \
-o custom_output.txtCommon flags:
--pick: One consequence per variant--per_gene: One consequence per gene--fork N: Use N CPU cores--no_stats: Skip HTML summary (faster)--tab: Explicitly requests tab-separated output with a header line suitable for downstream tools (R, pandas)--vcf: VCF format output--json: Outputs to JSON format--fields: Choose specific output columns
References
- McLaren W, et al. The Ensembl Variant Effect Predictor. Genome Biology (2016)
- Eilbeck K, et al. The Sequence Ontology: a tool for the unification of genome annotations. Genome Biology (2005)
- Richards S, et al. Standards and guidelines for the interpretation of sequence variants. Genetics in Medicine (2015)
- Terry A. Brown, et al. Genomes 5 (2023)